Содержание
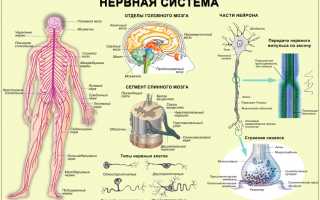
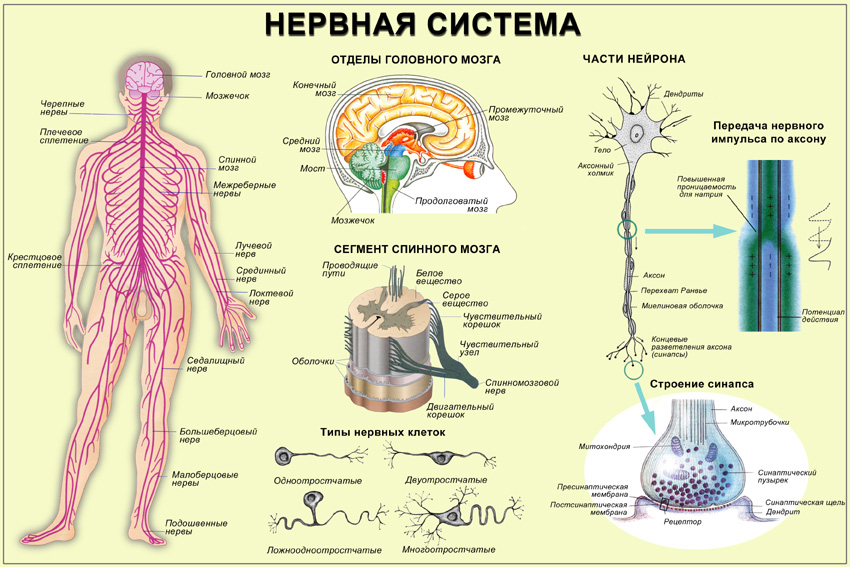
Наследственная нейрометаболическая патология, нарушающая обменные процессы и формирование жизненно важных компонентов центральной нервной системы (ЦНС) на уровне митохондрий, получила определение синдром Ли (инфантильная форма болезни Вернике). Аномалия относится к подострой некротизирующей энцефаломиелопатии, которая поражает серое вещество, а при хронической форме и белое, базальные ганглии, таламус, спинной мозг.
Заболевание обусловлено мутацией генов, проявляется в раннем возрасте с первой недели жизни до 3-х лет, наследуется по аутосомно-рецессивному или митохондриальному типу, поражает в равной мере оба пола. Характеризуется прогрессирующим клиническим течением с мышечной гипотонией, задержкой развития, эпилептическими приступами.
Классификация
Распределение разновидности синдрома Ли (болезнь Лея) зависит от того, на какой хромосоме находится «сломанный» ген, от дефектов белков в митохондриальной системе. Патологическому изменению, по которому определяется заболевание, подвержены Х-хромосомы, аутосомы и митохондриальная дезоксирибонуклеиновая кислота (ДНК) от первого по пятый отдел цепочки (I, II, III, IV, V). Виды поражений, связанные с мутацией:
- I комплекс (НАДН-KoQ) изменение ядерных генов: NDUFA10, NDUFS4, NDUFS3, DUFAF2. Дефекты в ДНК – причина MTND1,2, 3. В результате снижается выработка аденозинтрифосфата (АТФ), участвующего в энергетическом обмене на клеточном уровне. Передается двумя типами наследования.
- II (сукцинат-KoQ) характеризуется поражением белков из-за мутации на 5-й хромосоме SDHA. Ген контролирует активность субъединицы А сукцинат-KoQ- редуктаза, участвует в функции трикарбоновых кислот, дыхательной цепи, транспортировке электронов. Передается только по аутосомно-рецессивному типу.
- III (KoQН2-цитохром с-редуктаза), нарушение связаны с BCS1L, расположенном на 2 хромосоме. Понижение способности гена преобразовать наследственную информацию нуклидов в белок. Болезнь возникает на фоне дефективности протеина убихинон-с-редуктазы. Передается аутосомно-рецессивным путем.
- IV (цитохром с-оксидаза) изменения вызваны мутациями ядерных генов на 17 хромосоме SCO 1, COS10 и нарушениями митохондриальной ДНК. Пути передачи до конца неизвестны.
- V комплекс (АТФ-синтаза) характеризуется дефектом ATPAF2 (17 хромосома), что вызывает снижение окислительного процесса выработки аденозинтрифосфата.
Классификация включает болезнь Лея, обусловленную редкой формой мутации PDHA1, локализованного на Х-хромосоме. Аномалия распространяется только на мальчиков, носителем является мать. Девочка может наследовать «сломанный» ген, но сама болеть не будет, патология затронет ее потомство мужского пола.
Передача синдрома лея по митохондриальному типу наследования касается только женщин, они являются носителями дефектных клеточных органелл. Каждая митохондрия имеет свою ДНК, при делении яйцеклетки трудно определить распределение «больных» генов. Будучи фенотипически здоровой, женщина несет угрозу своему потомству.
Разовьется болезнь или нет, зависит от количества неполноценных митохондрий, полученных ребенком. Синдром распространяется только по женской линии, болеют как девочки, так и мальчики. Если ребенок мужского пола не получил достаточного количества мутированных генов для развития болезни, он не является угрозой для своего потомства.
Основные симптомы
К группе риска развития подострой некротизирующей энцефаломиелопатии относятся дети первого года жизни, в редких случаях заболевание проявляется в подростковом периоде. Клиническая характеристика синдрома лея начального этапа:
- вялое состояние, сонливость;
- возможна высокая возбудимость;
- грудные дети отказываются от питания (отсутствует глотательный и сосательный рефлекс);
- недобор стандартного для возраста веса.
Дальнейшее течение патологии сопровождается:
- непроизвольными движениями (миоклония);
- задержкой моторного и психического развития;
- слабостью мышц, сменяющейся гипертонусом;
- симптомами пирамидной недостаточности;
- нарушением кислотно-щелочного обмена (ацидоз);
- увеличением размера сердца, поражением миокарда;
- тремором;
- неполноценной вентиляцией легких, поверхностным дыханием;
- снижением рефлексов в области сухожилий;
- потерей координации движений.
Основной особенностью синдрома Ли является прогрессирующее течение. Эта форма патологии характеризуется явно выраженной симптоматикой:
 Читайте также:
Читайте также:

- тоническими судорогами, распространяющимися по всему телу;
- частичной или полной атаксией;
- нарушением зрительного нерва;
- парезами, потерей мышечной чувствительности;
- эпилептическими припадками;
- утратой приобретенных навыков;
- быстротекущим слабоумием (на фото).
Дефицит аденозинтрифосфата отражается не только на центральной нервной системе, но и на внутренних органах. У детей болезнь вызывает увеличение сердца, печени, развитие желтухи. В процессе прогрессирования патологии отмечается нарушение дыхательной функции, она приобретает симптомы Чейна-Стока (постепенное учащение), что, в свою очередь, вызывает гипервентиляцию легких и спазмы скелетной мускулатуры (тонические и клонические судороги). Примерно через 2–4 года наступает летальный исход из-за сердечной или дыхательной недостаточности.
Причины патологии
Этиологией прогрессирующего заболевания центральной нервной системы является мутация генов, кодирующих белки, которые участвуют в энергетическом обмене ряда комплексов, в том числе дыхательной цепочке митохондрий, в окислительном процессе производства АТФ. Клетка биологического организма содержит множество древних симбионтов с автономной ДНК, основной функцией которых является генерация жизненно необходимой энергии.
Окислительное фосфорилирование проходит в комплексах дыхательной цепи митохондрий, затем электроны никотинамидаденинди-нуклеотида (NАDН) транспортируются органеллами в межмембранный просвет. Чувствительные к энергетическому дефициту нейроглии клетки вызывают аномальные процессы в центральной нервной системе, как следствие, развиваются разнообразные нарушения в раннем детском возрасте.
В совокупности с генной мутацией причиной недостаточности фермента может послужить ряд факторов:
- заболевания ЦНС;
- опухоль или киста в мозжечке;
- образование глиальных элементов (заменителей поврежденных клеток) в головном или спинном мозге;
- процесс формирования новых сосудов, превышающих объем тканей (пролиферация);
- отмирание нейронов из-за воспалительных процессов;
- наследственная предрасположенность (генетическая детерминанта).
Причиной развития патологии может стать любая инфекция или некорректно проведенная вакцинация.
Диагностические исследования
Определение синдрома Ли не проводится в амбулаторных условиях, больного помещают в стационар. Начальный этап заключается в дифференцировании болезни от патологий с похожей клинической картиной:
- энцефаломиелитом;
- амавротической идиотией;
- инфекционно-аллергического генеза;
- миоклонус-эпилепсией;
- лейкодистрофиями.
При визуальном осмотре уделяется внимание неврологическим проявлениям:
-
Читайте также:

- тремор верхних и нижних конечностей;
- отставание в развитии;
- недостаток массы тела;
- фасцикуляция мышц языка.
Диагностика включает информативное изучение наследственного фактора, если заболевание имеет аутосомно-рецессивную форму. При мутации генов определить передачу патологии от матери ребенку невозможно. В этом случае рекомендуется молекулярный анализ.
Дальнейшие мероприятия проводятся с применением специального оборудования:
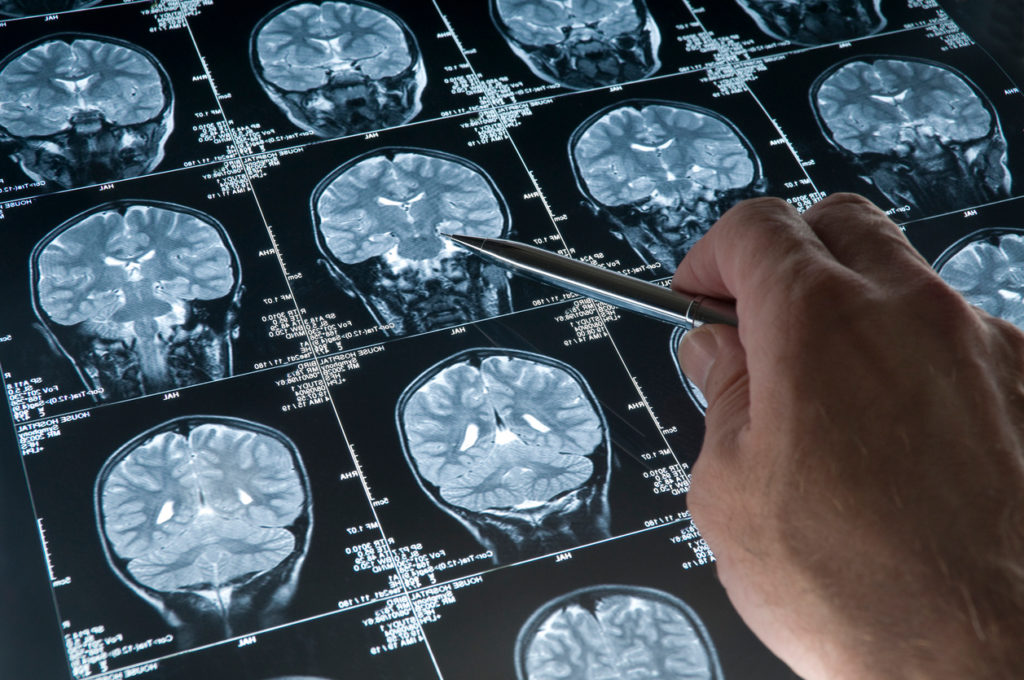
- Методом магнитно-резонансной томографии в FLAIR и T2 в этих режимах выявляют поражение продолговатого мозга, таламуса, базальных ганглиев, особенно чувствительных к гипоксии. Нарушения носят симметричный характер. В некоторых случаях наблюдается аномальное изменение в белом веществе полушарий, обусловленное кистозным новообразованием.
- Молекулярная диагностика применяется для выявления вида мутации SURF1 и ВCS1L гена.
- Электрокардиография.
- Офтальмоскопия глазного дна.
- Электронейромиография отображает разрушение миелина белого вещества периферической нервной системы (демиелинизацию) из-за снижения скорости прохождения импульсов. На ЭКГ фиксируется преобладание медленной активности волн.
Проводится забор спинномозговой ликвора для определения уровня белка и лимфоцитарного плеоцитоза. Путем биохимического исследования крови выявляется высокий показатель кетоновых тел, что свидетельствует о наличии лактат-ацидоза.
Эффективные методы лечения
Специальной терапии при некротизирующей энцефалопатии не разработано. Мероприятия направлены на улучшение работы ЦНР и носят симптоматический характер для стабилизации состояния ребенка. Лечение проводится применением таких медикаментов:
- антибактериального действия – «Цефтриаксон»;
- противогрибковые – «Флюконазол», «Дифлюкан»;
- противовирусные – «Завиракс»;
- антибиотики – «Биотин», «Ампициллин»;
- витамины – С, В1, Е 200, В6 («Пиридоксин»);
- для нормализации состояния – «Тиамин-хлорид», «Кокарбоксилаза»;
- глюкокортикостероиды – «Дексаметазон», «Преднизолон»;
- нейротропные средства – «Нейромидин», «Пирацетам»;
- устраняющие судороги – «Клобазам», «Эпилим», «Клоназепам»;
- гормональные препараты – «Гидрокортизон», «Тетракозактид»;
- для метаболической терапия – «Тиамин», «L-карнитин»; «Рибофлавин», «Коэнзим Q10», «Актовегин», «Церебролизин»;
- диуретики – «Фуросемид»;
- кардиометаболические средства – «Кардио – АБВ», «Милдронат»;
- муколитического действия – «Амброксол»;
- для нормализации микрофлоры кишечника – «Креон», «Линекс».
- препараты, в основе которых вальпроевая кислота.
При лечении патологии медикаментами рекомендуется соблюдение диеты, предусматривающей ограничение употребления продуктов с высоким содержанием белка, норма поступающего в организм фермента не более 1 гр в сутки. Если в семье есть ребенок с болезнью Лея, обязательна медицинская консультация для оказания первой помощи во время приступа.
Прогноз и рекомендации по профилактике
После зачатия, если есть предпосылки генетического наследия, актуально провести перинатальную диагностику плодного материала. Необходимость исследования заключается в выявлении анеуплоидий синдрома Дауна, Ли, Шерешевского-Тернера. Если результат окажется положительным, беременность прерывается. У детей с болезнью Лея продолжительность жизни составляет от 2 до 5 лет. Симптоматическая терапия замедляет прогрессию патологии, а не избавляет от нее. При хронической форме нарушения приводят к инвалидизации и летальному исходу от сердечной и дыхательной недостаточности. Профилактика патологии заключается в медико-генетической консультации обоих родителей перед планированием беременности.