Содержание
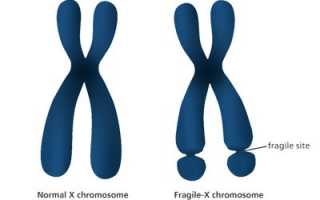
В 40-х годах двадцатого века учеными Джеймсом Мартином из Ирландии и англичанкой Джулией Белл впервые описана клиническая картина заболевания. Генетики на протяжении определенного времени наблюдали семью, где у абсолютно нормальной женщины рождались умственно отсталые мальчики. Отследив фамильную историю, выявили прецеденты у мужчин в предыдущих поколениях. Ученые установили причину генетической аномалии, заключавшуюся в ломкости дистального плеча X-хромосомы. Визуально отмечалось сужение концов за счет вторичной перетяжки на истонченном участке в локусе Xq27-28 (фото).
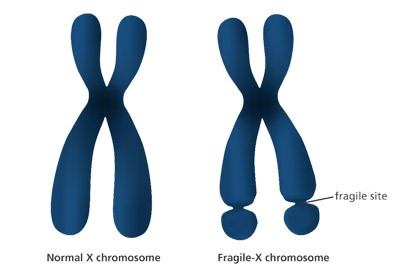
В 90-х годах путем цитогенетического обследования был выявлен мутированный ген, послуживший причиной синдрома фрагильной хромосомы. В название патологии вошли имена первых ученых, обративших внимание на изменение этого типа в геноме. Заболевание, передающееся по наследству, сцеплено с полом. У мальчиков симптоматика явно выражена, у девочек встречается значительно реже, протекает в легкой форме умственной отсталости. Относится к распространенной аномалии (1:4000), по частоте возникновения занимает ведущее место среди наследственных патологий.
Причины возникновения
Генотип человека составляют 46 хромосом, две из них определяют пол – X, Y. У женщин набор 46 XX, у мужчин – 46 XY. Этим объясняется редкое проявление болезни у девочек, когда происходит компенсация со стороны второй хромосомы кариотипа. Цепочка генетической памяти состоит из повторяющихся комбинаций цитозин-гуанин-гуанин (ЦГГ), увеличение копий (экспансия) истончает участки ДНК, вызывая Синдром Мартина-Белла (СМБ). Аномалия возникает на фоне мутации FMR1, гена, отвечающего за кодировку белка, основного участника формирования нервной системы.
Отрезки X хромосомы характеризуются четырьмя категориями
| Состояние | Показатель чередования тринуклеотидов | Развитие болезни |
|---|---|---|
| нормальное | 29–30 | отсутствие СМБ |
| промежуточное (серая зона) | 44–55 | риск развития |
| премутация | 60–200 | синдром не развивается, человек является носителем сломанного гена, болезнь проявится в следующих поколениях |
| полное нарушение цепочки | 250–4000 | появление аномалии |
Мутация гена угнетает функциональность белка, участвующего в развитии ребенка, влияющего на его способность к обучению и запоминанию нового материала. Недостаточность фермента сказывается на формировании аксонов, синапсов, принимающих непосредственное участие в нервных связях, на этом фоне развиваются неврологические отклонения и умственная отсталость.
Наследуется генетическая патология по женской линии. У мужчины, имеющего одну X-хромосому, после передачи сломанного гена от матери дебют синдрома приходится на двенадцатый месяц жизни с последующей прогрессией. Передать аномалию он может только дочери. У девочки дефицит пополняется за счет второй хромосомы. Патология не проявляется, в худшем случае сопровождается легкой симптоматикой. Женщина передает синдром ломкой Х-хромосомы потомству обоих полов, круг замыкается. Поэтому в пределах одного родового клана у мужчин отмечается нарушение умственного развития, при этом женская половина совершенно здорова или с незначительными отклонениями.
Характерные признаки патологии
Генетическое заболевание сопровождается разнообразием признаков, каждый случай индивидуален набором собственной симптоматики. СМБ отличает от других неврологических патологий ряд особенностей: расстройство психоэмоционального восприятия, прогрессирующая умственная отсталость, отклонение в физическом развитии. Синдром Мартина-Белла у детей характеризуется симптомами, позволяющими без труда определить форму генетической мутации. Новорожденный мальчик отличается большим весом и увеличенным размером яичек (макроорхизм), без гормональных отклонений.
Отмечается снижение сосательного и хватательного рефлекса, мышечного тонуса, он плохо реагирует на внешние раздражители. Ребенок отстает от сверстников в физическом и интеллектуальном развитии. В большинстве случаев появляется на свет с врожденными заболеваниями: пороками сердца, деформацией суставов. Поэтому дети с СМБ поздно начинают ходить, практически не ползают, речевая функция носит заторможенный характер, словарный запас скудный, дикция невнятная, в тяжелых случаях проявляется полное отсутствие коммуникативных способностей, ребенок молчит.
По мере прогрессии синдрома Мартина-Белла признаки дополняются, имеют более выраженный характер. При задержке психомоторного развития движения приобретают гиперкинетические особенности:
- бессистемное размахивание руками, хлопки в ладоши, встряхивание фалангами;
- прыжки на месте;
- круговые движения туловищем, повороты вокруг своей оси;
- дискоординация, бессмысленность и помпезность поз.

 Читайте также:
Читайте также:
Психологические отклонения:
- эмоциональная лабильность (реакция на раздражитель не соответствует возрасту);
- бесконтрольное проявление гнева, агрессии;
- необоснованное упрямство, плаксивость, дефицит внимания;
- боязнь физического контакта, скопления малознакомых людей, громких звуков;
- симптомы аутизма.
Неврологические аномалии, свойственные синдрому:
- эпилептические приступы на фоне мышечных судорог, временной потери сознания;
- нервный тик, локализованный в нижней части лица и веках, вызывающий искривление мимики и частое моргание;
- тремор верхних конечностей;
- гиперподвижность, больной неспособен находиться на одном месте длительное время:
- глазодвигательные, пирамидные расстройства.
Синдром фрагильной хромосомы формирует физические отклонения в развитии, больные мальчики визуально отличаются от здоровых сверстников:
- голова большого размера с выпуклым высоким лбом, от этого лицо приобретает форму овала;
- небный свод глубокий, нижняя челюсть тяжелая;
- уши круглые, оттопыренные, с низкой посадкой на черепе;
- нос заостренный, в виде крючка;
- глаза широко поставлены, косят.
Фенотип дополняется эластичностью кожи, плоскостопием, кривизной ног, широкими ступнями и кистями.
У всех носителей сломанного FMR1 неизменной симптоматикой является дисфункция щитовидной железы, надпочечников. Эндокринные нарушения вызывают недостаточность метаболизма (ожирение), раннее половое созревание. Уровень умственной отсталости колеблется от легкой формы до тяжелого клинического течения. Основной процент пациентов находится на стадии олигофрении.
Женщины отличаются высоким либидо, но у них наступает ранний период менопаузы. Происходит перерождение яичников в кистозные новообразования. У мужчин отмечается явный макроорхизм.
Диагностические исследования
Определение синдрома Мартина-Белла предусматривает применение специфических тестов для анализа состояния Х-хромосомы на Xq27-28 участке. Проводится врачом генетиком по следующему алгоритму:
- Осмотр пациента с учетом специфических изменений внешности и гипотонуса мышечной массы.
- Основной методикой в диагностике патологии, дающей 100% результат на раннем этапе клинического развития, является цитогенетический способ. Берутся клетки больного, обрабатываются фолиевой кислотой, которая запускает процесс изменения хромосомы. Если аномалия отметилась на Xq27-28 лакмусе, наличие синдрома не вызывает сомнений.
- На более поздних сроках применяется исследование пары хромосом, отвечающих за пол (кариотипирование). Мутация подтверждает СМБ.
- При помощи полицепной реакции анализируется состав и структуру тринуклеотидов.
- Молекулярно-генетическое изучение определяет частоту повторов ЦГГ.
- При синдроме ломкой X-хромосомы у всех пациентов одинаковая биоэлектрическая активность головного мозга, что позволяет при помощи электроэнцефалографии подтвердить диагноз.
Болезнь можно выявить на ранних стадиях беременности. Перинатальное обследование основывается на УЗИ, анализе сыворотки крови женщины, биопсии хориона. Если подтвердилась генетическая аномалия у плода, предлагается прерывание беременности, но в любом случае выбор остается за будущей матерью.

-
Читайте также:

Эффективное лечение
Как и от любого генетического заболевания, передающегося по наследству, от синдрома Мартина-Белла избавиться невозможно. Терапия медикаментами проводится комплексно с физиопроцедурами, в крайних случаях прибегают к оперативному вмешательству. Мероприятия направлены на уменьшение симптоматики и призваны улучшить качество жизни, предотвратить прогрессирование умственной отсталости и неврологических отклонений.
Консервативные методы
Лечение генетической аномалии предусматривает применение таких препаратов:
- разжижающих кровь – «Клексан», «Плавикс»;
- предупреждающих эпилептические приступы – «Мазепин»;
- ноотропного действия – «Пирацетам»;
- для улучшения состояния сосудов и мозгового кровообращения – «Церебролизин», Винпоцетин»;
- седативного (успокаивающего) эффекта – «Седуксен», «Диазепам»;
- антидепрессантов – «Сертралин», «Флуоксетин», «Кломипрамин»;
- соматического действия (психостимуляторы) – «Солкосерил», «Кавинтон», «Лидаза»;
- нейролептиков – «Хлорпромазин», «Галоперидол», «Перициазин».
В комплексной терапии применяют препараты на основе лития вместе с набором витаминов, которые нормализуют когнитивную функцию. Стремление лечить синдром фолиевой кислотой оказалось неэффективным. Терапия на время улучшала поведенческие и коммуникативные возможности, но не тормозила процесс умственной деградации.
Физиотерапия
В помощь консервативному воздействию на проявления синдрома назначается ряд физиотерапевтических мероприятий:
- лечебная физкультура;
- упражнения в бассейне;
- душ Шарко;
- грязевые ванны с радоном;
- акупунктура (иглоукалывание);
- гирудотерапия (пиявки для разжижения крови);
- мышечная релаксация.
Показаны занятия с логопедом, тренинги с психотерапевтом.
Оперативное лечение
Хирургическое вмешательство целесообразно, если осложнение синдрома Мартина-Белла затронуло жизненно важные органы. Делается операция при врожденных пороках сердца, кистозном перерождении яичников с риском перехода в злокачественное образование.
Применяется пластическая коррекция, цель которой – устранение физических дефектов, свойственных болезни. Хирургическим методом приводятся в норму конечности, меняется форма ушей, устраняется внешняя аномалия половых органов.
Прогноз и профилактика
Генетическая ломкость X-хромосомы не создает больших проблем со здоровьем, если не осложнена патологиями внутриутробного развития. Продолжительность жизни не отличается от здоровых людей. Прогноз на выздоровление неблагоприятный. При адекватном симптоматическом лечении, психологической коррекции, помощи человеку с адаптацией в социуме качество жизни значительно улучшится, но в итоге синдром приведет к инвалидности.
Профилактикой заболевания является перинатальное обследование плода на ранних сроках. Скрининг биологического материала поможет выявить мутацию на Xq27-28 участке. В этом случае рекомендуется прерывание беременности. Мужчине или женщине, в роду которых были прецеденты мутации FMR1, перед планированием ребенка необходимо пройти тест. Если аномалия у одного из родителей подтверждается, на молекулярном уровне существуют способы исправить дефект X-хромосомы и провести экстракорпоральное оплодотворение. ЭКО даст возможность рождения младенца со здоровым генетическим кодом.
-
Читайте также: