Содержание
Новозеландский кардиолог Джон Вильямс, наблюдавший детей с пороками сердечно-сосудистой системы, обратил внимание на одинаковый фенотип нескольких пациентов. В 1961 году в своих работах доказал врожденную природу заболевания, которое получило название синдром Вильямса-Бойрена (Уильямса), «лица эльфа».
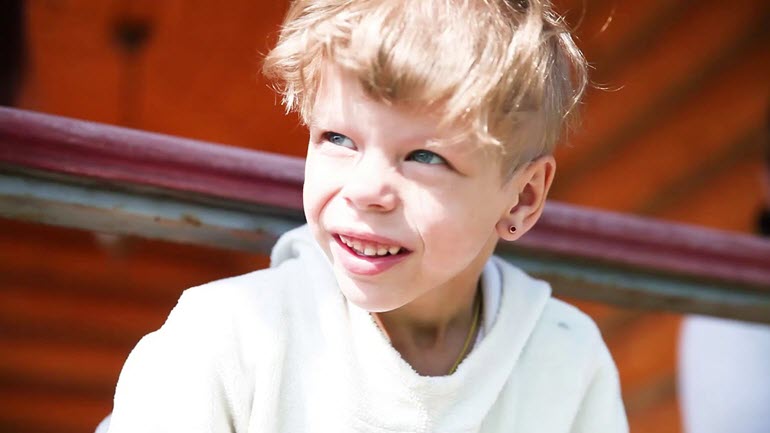
В основе развития болезни лежит отсутствие нескольких генов в хромосомной цепочке. Отличительной чертой является специфическое строение лица. Дети с заболеванием имеют необычную внешность (на фото), обладают уникальной особенностью развития интеллекта в определенной сфере на фоне умственной отсталости. В редких случаях патология передается аутосомно-доминантным способом.
Факторы-провокаторы синдрома Вильямса
В научных кругах ведутся споры вокруг причины, вызывающей генетическое заболевание. Часть специалистов поддерживает теорию наследственного фактора, остальные склоняются к спонтанной мутации во время зачатия. Механизмом, запускающим патологический процесс, является делеция (утрата) пары в седьмой хромосоме. В состав отсутствующей копии входит больше 25 кодирующих генов, от которых зависит:
- углеводный метаболизм;
- синтез эластина;
- функционирование головного мозга;
- кальциевый обмен;
- состояние нейронов в нервной ткани;
- развитие интеллекта.
Недостаточное производство эластина, входящего в клеточный состав стенок сосудов и сердечного клапана, приводит к появлению пороков органа и гипертензии. Нарушение метаболизма кальция становится причиной гиперкальциемии, при которой истончаются стенки артерий и клапана, вызывая сердечно-сосудистую недостаточность. Дефицит питания нейронов отражается на формировании центральной нервной системы, развитии головного мозга, лицевой дисморфологии.
Вероятность синдрома Вильямса у потомства возрастает, если генетическая мутация диагностировалась у родителей или близких родственников. В этом случае не отмечено выпадение одной копии в наборе на момент зачатия. С учетом клинических проявлений у новорожденного недостающее звено в седьмой хромосоме плод получил путем наследования. Такая причина развития аномалии встречается очень редко.
Более распространенная теория, чем аутосомно-доминантный вариант, утверждает, что синдром Вильямса у детей формируется путем спонтанного нарушения хромосомной цепочки в момент зачатия. Провоцирующим фактором является воздействие на организм одного или обоих родителей:
- вида трудовой деятельности на вредном производстве, связанном с ядовитыми химикатами, тяжелыми металлами, радиацией;
- концентрация токсинов в организме по вине плохой экологии в месте проживания мужчины или женщины;
- употребление алкоголя, наркотиков, курение табака.
В этом случае у родителей существует риск появления на свет ребенка с недостающим звеном в хромосоме.

Отличительные признаки аномалии
К основным визуальным симптомам заболевания, которые определяются в младенческом возрасте, относятся специфические черты лица и строение черепа. Благодаря этой особенности, аномалия получила свое второе название – синдром «лица эльфа». В процессе развития ребенка видоизменяется скелет и мышечная структура. Характерные особенности внешности:
 Читайте также:
Читайте также:

- высокий, широкий лоб, непропорциональный зауженному книзу маленькому подбородку;
- плоская переносица переходит в короткий нос, у которого отсутствует кончик;
- разрез широко посаженных глаз узкий, цвет зрачка голубой, веки припухшие, сходящееся косоглазие;
- низко расположенные уши вытянутой кверху формы, что делает индивидуума похожим на персонажа фэнтези;
- рот большой, верхняя губа значительно тоньше нижней.
Кукольную внешность дополняют пухлые опущенные щеки, прямые, правильной формы брови.
У детей с этим диагнозом позже установленного нормой срока прорезаются зубы, деформируется челюсть, проявляются признаки неправильного прикуса. Отсутствие аппетита сказывается на росте, отмечается дефицит веса. Мышцы недостаточно развитые. К вторичной симптоматике синдрома относится плоскостопие, слабый вестибулярный аппарат, нарушение координации.
У человека в переходном возрасте признаки дополняются:
- заниженной линией талии;
- узкой грудной клеткой;
- непропорциональной туловищу длинной шеей;
- соединенными коленями.
Дефекты со стороны внутренних органов характеризуются:
- Выпячиванием внутрь стенки двенадцатиперстной кишки, что становится причиной частых запоров. Заболевание сопровождается хроническим гастритом.
- Формированием стеноза артерий и аорты, сердечной недостаточностью. Избыток кальция в организме усугубляет патологию.
- Недоразвитость щитовидной железы у плода становится причиной гормонального сбоя, появляются признаки сахарного диабета, гипотиреоза.
- Аномалией почек и мочевыделительной системы.
У всех пациентов болезнь вызывает отставание в умственном развитии. Дети неспособны образно мыслить, не могут сконцентрироваться на определенном предмете, у них отмечается дефицит внимания.
Малыши плохо читают, практически не владеют счетом. Невозможность обучения компенсируется хорошим словарным запасом, высокими коммуникативными способностями. В возрасте до четырех лет пациенты испытывают трудности в составлении осмысленного предложения, позже эта проблема самоустраняется.
Малыши в частых случаях обладают абсолютным слухом и чувством ритма. Легко знакомятся с людьми, у них хорошая память на лица. Привлекают внимания обширным словарным запасом и умением им пользоваться. Легко ведут беседу, доброжелательны, всегда позитивно настроены, улыбчивы и вежливы. Синдром Уильямса делает ребенка знаменитостью в кругу социума.
Взрослые отличаются инфантильностью, которая проявляется в несвойственных возрасту поступках, гиперактивностью, эмоциональной лабильностью. Необходимость в постоянном общении вызывает неадекватность поведения, формирует фобии. Для адаптации в социуме пациентам необходима помощь психолога и близких людей.
Диагностические мероприятия
Определение синдрома Уильямса ведется в нескольких направлениях, назначаются лабораторные исследования, инструментальные методики, учитывается симптоматика. Достоверная диагностика, с высокой вероятностью показывающая патологию, – это генетическое тестирование. Хромосомная перестройка выявляется путем проведения флуоресцентной гибридизации, указывающей на последовательность генов. Молекулярно-цитогенетический анализ осуществляется ДНК-микрочипом, который вводится во время беременности и после рождения ребенка.
Лабораторные методики, включающие общий и биохимический анализ крови, устанавливают концентрацию кальция, уровень тиреотропного гормона. Показатели этих веществ при заболевании будут повышены, а количество нейтрофилов в сыворотке, наоборот, понижено. Проводят исследование мочи для выявления креатинина и вычисляют соотношение показателя с концентрацией кальция. Генетическая мутация этого типа всегда сопровождается гиперкальциемией и гиперхолестеринемией.
-
Читайте также:

Инструментальная диагностика:
- Тонометрия нижних и верхних конечностей для определения в них артериального давления (ярко выраженная гипертензия).
- ЭхоКГ (кардиологическое исследование) для выявления врожденных аномалий сердца и сосудов. При синдроме отмечается недостаточность сердечных клапанов, стеноз аорты и крупных артерий.
- УЗИ (ультразвуковое исследование) почек.
- Томография головного мозга.
- Рентгенография урологических органов.
Пренатальный скрининг плода позволяет определить пороки в сердечно-сосудистой системе, поликистоз почки. На ранних сроках беременности помогает выявить пониженную концентрацию в крови женщины альфа-фетопротеина. Своевременно проведенная диагностика способствует назначению адекватной терапии. При необходимости проводится хирургическое вмешательство. Квалифицированная помощь, оказанная новорожденному, в дальнейшем улучшает качество и продолжительность жизни.
Принципы лечения
Учитывая, что синдром «лица эльфа» относится к генетическому заболеванию, вызванному перестройкой в хромосоме, этиотропного лечения еще не разработано. Задачей терапии является коррекция физических аномалий, интеллектуального развития. Медикаментозное воздействие направлено на устранение или облегчение симптомов. Назначаются следующие препараты:
- Ноотропные средства – «Пирацетам», «Семакс», «Винпоцетин», «Церебролизин», «Актовегин», нормализующие кровообращение в коре головного мозга.
- Гипотензивные препараты, купирующие высокий показатель артериального давления, антагонисты кальция, мочегонные медикаменты, блокаторы бета-адринолина рецепторов.
- Если синдром протекает в тяжелой форме, применяются глюкокортикостероиды «Преднизолон», «Гидрокортизон».
- Средства седативного влияния – «Валериана», «Ново-пассит». При выраженной гиперактивности рекомендуются сильнодействующие лекарства.
- Для нормализации высокого уровня кальция необходима внутривенная инъекция физраствора с препаратами калия.
В комплексную терапию включаются: лечебная физкультура, массаж, иглоукалывание, электро- и магнитотерапия. Оперативное вмешательство показано для устранения врожденных пороков сердца.
Психологическая помощь улучшает способность к обучению, освоению навыков общения, адаптации в коллективе. Для детей с «лицом эльфа» необходимо создать дружелюбную обстановку в семье, не ограничивать контакт со здоровыми сверстниками, благоприятные факторы помогут снизить выраженность задержки умственного развития.
Прогноз и профилактика
Специальных мер, предотвращающих появление синдрома, нет. В большинстве случаев генная мутация происходит спонтанно, без видимых для этого причин. Для исключения прогрессирования врожденных патологий рекомендуется:
- периодически отслеживать концентрацию холестерина и кальция в крови;
- отказаться от лекарств и продуктов, содержащих витамин D;
- рацион не должен провоцировать запоры;
- систематические физические нагрузки помогут избежать излишка веса при гормональном дисбалансе;
- достаточное время на ночной сон и дневной отдых уменьшает излишнюю активность, препятствует появлению тревоги;
- в случае неоднократного повышения температуры тела необходимо обследовать ребенка на локализацию инфекции в мочевыделительной системе;
- занятия проводят в спокойной дружелюбной обстановке, предварительно убрав предметы, способные отвлечь внимание.
Рекомендуется проходить плановую диспансеризацию, включающую консультирование кардиолога, уролога, психотерапевта, ортопеда.
Прогноз синдрома Вильямса и продолжительность жизни пациента зависят от степени врожденных дефектов. Выздороветь человек полностью не сможет, проведенная в раннем возрасте операция по устранению пороков даст возможность дожить до преклонной старости. Прогноз улучшается, если проводится регулярный мониторинг состояния сосудов и сердца. Мероприятие позволит избежать артериальной гипертензии и сердечной недостаточности, причины летального исхода.
-
Читайте также: