Содержание
В начале двадцатого века на заболевание обратил внимание немецкий врач В. Брахман. На примере наблюдаемого пациента он описал основные симптомы генетической патологии. Позже, в тридцатых годах того же столетия, голландский педиатр Корнелия де Ланге (де Лонга) в разное время вела пятерых пациентов, не состоящих в родстве и имеющих сходные признаки заболевания. По итогам исследования врач сделала заключение и опубликовала работу.
Синдром относится к редким генетически гетерогенным болезням, в равной степени поражает девочек и мальчиков, встречается на всех континентах. По статистическим данным, на 10–30 тысяч новорожденных приходится один случай с этой патологией. Еще одно название синдрома – Амстердамский нанизм (карликовость). В городе зафиксировано три индивида с врожденной аномалией. Болезнь протекает на фоне пороков развития скелета, внутренних органов, умственной отсталости.
Причины возникновения
Патогенезом аномалии является мутация генов NIPBL, SMC3, SMCIA, их белки участвуют в работе когезинового комплекса, присоединяя фермент к хромосоме в метафазе. Нарушение функционирования лежит в молекулярной основе синдрома. У половины пациентов определяется сломанный генетический код NIPBL, содержащий больше 40 экзонов, гетерозиготные мутации вызывают сбой в процессе de novo.
Были зафиксированы случаи передачи синдрома аутосомно-доминантным путем. Мозаичная форма NIPBL отмечалась у родителей в стадии эмбриона. Носители сломанного гена способны передать его потомкам через одно или через несколько поколений. У совершенно здоровой женщины и мужчины возможно рождение ребенка с синдромом Корнелии де Ланге.
В пяти процентах случаев аномалия формируется по вине мутации SMC1A, он также кодирует белки, участвующие в когезиновом процессе. Заболевание характеризуется легкой формой фенотипа. Нарушенный ген представляет опасность для всех поколений семьи, потому что напрямую связан с Х-хромосомой. Описан один пациент с изменением SMC3 с характерными чертами лица и строением черепа, при этом недоразвитость внутренних органов отсутствовала. В 40% случаев этиологию еще предстоит выяснить.
Обширный перечень симптомов
Определить наличие аномалии можно визуально сразу после рождения младенца. Ребенок появляется на свет с дефицитом веса (2/3 от общепринятой нормы). Синдром Корнелии де Ланге у пациента проявляется отличительными чертами в строении скелета, сопровождается неврологическим расстройством, отсталым умственным развитием.
Аномалии головы
Отклонение от критериев в строении лица и черепа является основным показателем для определения патологии.
Отмечается:
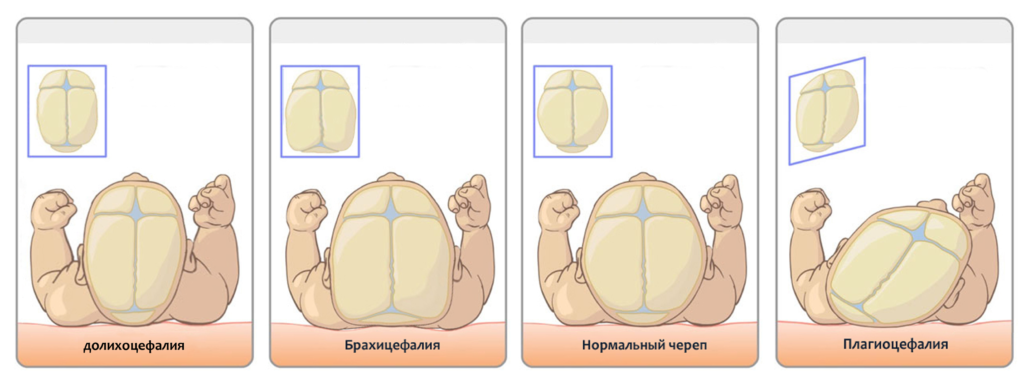
- объем меньше на 15%, чем у здорового новорожденного (микроцефалия);
- ширина больше продольного размера (брахицефалия);
- верхняя (мозговая) часть уменьшена.
Дефекты лица:
- четко очерченные брови, сросшиеся (сплошная линия), негустые, недоразвитые надбровные дуги;
- ресницы длинные, пушистые, загнутые кверху, разрез глаз – по монголоидному типу;
- переносица запавшая, широкая, нос маленький, с явно выраженными ноздрями;
- нижняя челюсть не пропорциональна лицу, маленькая, квадратной формы;
- губы тонкие, с опущенными уголками;
- редкие зубы (олигодонтия) или полное их отсутствие;
- небо высокое, без перегородки (волчья пасть).
В 50% случаев выявляется повышенная волосистость тела в основном в области спины, низкая линия роста волос на лбу и затылке. Кожный покров имеет синюшный оттенок, хорошо просматривается сетка сосудов (мраморность). Последний признак не относится к основным показателям синдрома.
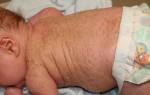
Недоразвитость внутренних органов
Аномалии внутренних органов являются серьезной угрозой для жизни индивида. При заболевании отмечается:
- закрытость носовых проходов (атрезия хоан), пациент дышит при помощи зонда;
- пороки со стороны сердца: дефекты в структуре сосудистой системы, перегородок, клапана, отверстие в предсердии;
- нарушения желудочно-кишечного тракта: сужение привратника, подвижность слепой кишки, грыжа на диафрагме, желудочный рефлюкс, стеноз пищевода;
- аномалии развития в мочевыводящей системе;
- недоразвитость половых органов: маленький половой член, пустая мошонка (отсутствие яичек), крипторхизм, двурогая матка;
- новообразование на почках, гидронефроз.
Также патологию сопровождают дефекты в мозговой ткани, характеризующиеся аплазией мозолистого тела и дисплазией извилин.
Костно-мышечный аппарат
При синдроме отмечается патологическое строение конечностей:
- укороченные руки по отношению к телу, отсутствие предплечий;
- неподвижность локтевого сустава (анкилоз) в результате образования хряща;
- сращивание нескольких пальцев (синдактилия), искривление пятого (клинодактилия);
- фаланги квадратной формы, приплюснуты;
- фокомелия, развитые ступни и кисти на фоне микромелии (короткие руки и ноги);
- отсутствие бугорков мышц на мизинце и большом пальце;
- образование глубоких складок на ладони;
- нижние конечности подвержены аналогичным изменениям.
Дефекты в развитии позвоночника, рост ниже нормы, соответствующей году ребенка, деформированная грудная клетка, короткая шея.
Неврологические отклонения
Проявление болезни характеризуется следующими нарушениями нервной системы:
- косоглазие, близорукость, атрофия глазного нерва, птоз (опущение верхнего века);
- гипертонус мышц, судороги;
- нарушение в работе сегментарного аппарата, которое приводит к повышению рефлексов;
- расположение рук на уровне глаз;
- хождение по кругу;
- нарушение сна;
- частичная потеря слуха или глухота;
- склонность к самоповреждению, депрессии.
У ребенка с возрастом развивается синдром дефицита внимания, он не может длительное время заниматься одним делом, концентрация отсутствует, проявляются признаки гиперактивности, отмечается невроз навязчивых состояний.
Умственное развитие
Неврологические отклонения и аномальные изменения в головном мозге влияют на умственное развитие, способствуют проявлению имбецильности. В легкой форме патологии пациент способен к обучению, словарный запас находится в рамках бытового общения. Такие индивиды посещают детские учреждения, но при этом не обладают критическим мышлением, трудно сходятся со сверстниками, возможно проявление аутизма.
Более тяжелая форма сказывается на коммуникативных способностях. Адаптация к социуму проходит в специализированных учреждениях. Склонность к проявлению агрессии. Невозможность самостоятельного существования в незнакомой обстановке. Пациенты неспособны к самообслуживанию, требуют ухода со стороны близких людей. Тяжело поддаются обучению. С годами процесс деградации только усугубляется.
 Читайте также:
Читайте также:

Диагностика
Определить наличие болезни у эмбриона при помощи современной диагностики практически невозможно. Показателем развития синдрома является низкая концентрация в плазме крови РАРР-А, этот вид протеина у женщины в период вынашивания ребенка производится в большом количестве. Обнаружить хромосомное отклонение у плода можно только с условием уменьшения показателя РАРР-А. Поэтому определяется патология после рождения младенца по визуальным признакам.
При наличии хотя бы одного симптома назначается инструментальная диагностика для выявления дефектов развития внутренних органов, несовместимых с жизнью. Методика включает:
- ультразвуковое обследование;
- магниторезонансную томографию;
- риноскопию;
- рентгенографию.
Назначаются клинические и цитогенетические анализы.
Необходим дифференциальный подход, чтобы исключить синдром, похожий на Корнелия де Ланге. Тяжелый фенотип имеет сходство с проявлением аутосомно-рецессивным заболеванием Фринса, которому свойственны такие признаки: грыжа диафрагмы, изменение лица, волчья пасть, недоразвитые конечности. Различием является заячья губа и макростомия (большой вес плода). Новорожденный с алкогольным синдромом также внешне похож на больного с Амстердамским нанизмом. Это объясняется задержкой внутриутробного развития. Различием является нормальный размер конечностей и расположение фаланг. В этом случае установить диагноз поможет семейный анамнез и хромосомный анализ.
Продолжительность жизни
При синдроме Корнелии де Ланге продолжительность жизни зависит от степени дефекта развития внутренних органов. Если аномалия несовместима с жизнью и своевременно не проведено оперативное вмешательство, смерть может наступить в течение месяца после рождения. Отсутствие соматических отклонений, ранняя диагностика дефектов, позволяющая их устранить, обеспечивает долгую жизнь пациентам. Прогноз ухудшается в случае вирусных инфекций, которым неспособна противостоять иммунная система. Обычный грипп может закончиться летальным исходом. В среднем дети с синдромом Корнелии де Ланге доживают до 13 лет. Фиксировались случаи при легкой, стертой форме, после адекватного устранения аномалий, когда пациент умирал в преклонном возрасте.
Лечение и профилактика синдрома Корнелии де Ланге
При офтальмологических дефектах проводится хирургическая коррекция птоза, лечение конъюнктивы, ношение очков. Рекомендуется своевременное купирование отитов стандартным набором антибиотиков, устранение заболеваний среднего уха, установка слухового аппарата. Пороки сердца удаляются оперативным путем. Аномалии, связанные с почками и мочевыделительной системой, убираются традиционными консервативными методами.
Профилактические меры
С учетом неточной картины патогенеза рекомендации, предупреждающие заболевание, призваны не допустить наследственной передачи мутированного гена. Профилактические мероприятия заключаются:
- в обязательном обследовании беременной женщины на уровень протеина-А в крови;
- не допускать инфекционных заболеваний особенно в период первого триместра;
- предотвращение зачатия путем инцеста (родители ребенка – кровные родственники);
- постоянное наблюдение у врача беременной женщины после сорока лет, обследование мужчины в случае позднего отцовства.
Рекомендуется медико-генетическая консультация перед зачатием родителю, у которого в семейном анамнезе есть прецеденты синдрома Корнелии де Ланге.
-
Читайте также: